HERCULES Web Server Documentation
HERCULES (Hybrid framEwoRk for RNA-binding domain loCalization and mUtation anaLysis using physicochemical and languagE modelS) is a sequence-based deep learning integrative method that predicts whether a protein can bind RNA, identifies RNA-binding regions at residue-level resolution, and estimates how single or multiple amino-acid changes affect RNA-binding propensity.
Method Description
HERCULES integrates two complementary sequence-only components. First, a protein language model (ProteinBERT) (1) is fine-tuned on a curated binary dataset of human RNA-binding proteins (RBPs) versus non-RBPs (2-8). This module outputs a global RNA-binding propensity score and, by extracting and averaging attention values across heads and layers, produces a residue-level signal that highlights regions contributing to RNA binding. This attention-derived profile captures long-range and context-dependent information such as domain architecture and sequence-wide constraints.
Second, HERCULES includes an explicit residue-level physicochemical module designed to be sensitive to local biochemical perturbations, particularly those induced by point mutations. For each sequence it computes 82 physicochemical descriptors at residue resolution (from established descriptor sets, including cleverSuite (9) and catGRANULE 2.0 ROBOT (7)). Feature selection is performed with an elastic-net regularized logistic regression, retaining 30 informative descriptors enriched in known determinants of RNA binding such as charge, hydrophobicity and disorder-related properties. Mutation effects are modeled by pairing wild-type and mutant sequences, computing WT-mutant descriptor differences, and learning an optimal linear combination of the selected features using Fisher's Linear Discriminant Analysis, yielding a mutation-sensitive physicochemical signal.
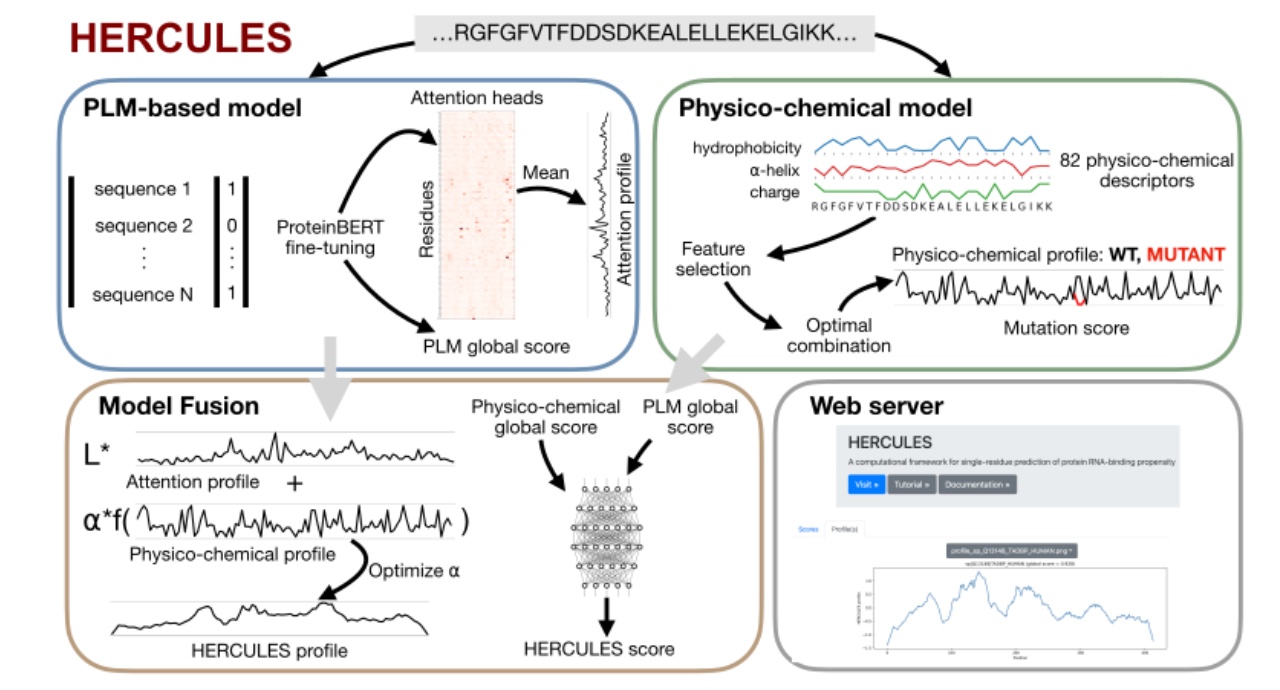
Figure 1. Figure 1. Overview of the HERCULES framework for sequence-based prediction of RNA-binding propensity. A protein language model (PLM; ProteinBERT) is fine-tuned to discriminate RNA-binding proteins (RBPs) from non-RBPs, yielding a global RNA-binding score and residue-level profiles obtained by averaging attention heads. In parallel, a physicochemical model computes 82 residue-level descriptors, performs feature selection, and predicts mutation-sensitive effects based on experimentally validated RNA-binding–disrupting variants. The two residue-level profiles are combined through an optimized linear weighting to produce the final HERCULES RNA-binding profile. Global scores from the PLM and the mean physicochemical profile are integrated by a multilayer perceptron to generate a fused global RNA-binding score. The HERCULES web server outputs include the residue-level profile, the RNA-binding global score and a mutagenesis heatmap.
Residue-level Profile Generation
The final HERCULES residue-level profile is obtained by fusing the language-model attention profile with the physicochemical profile. Because ProteinBERT attention values scale with protein length, the attention profile is multiplied by sequence length to make scores comparable across proteins. The physicochemical profile is smoothed with a sliding window and combined with the length-scaled attention using a weighted sum:
The hyperparameters a and l are optimized to improve separation of Pfam-annotated RNA-binding domains and to increase performance in identifying experimentally validated RNA-binding-disrupting mutations. Fused profiles are then z-score normalized using global normalization parameters estimated from merged training and test sets of the binary classification task. The result is a normalized, smoothed per-residue RNA-binding propensity track, referred to as the HERCULES profile.
Global RNA-binding Propensity Score
In addition to residue-level predictions, HERCULES outputs a global RNA-binding propensity score (the HERCULES score). This score integrates two complementary global signals: the ProteinBERT fine-tuned classifier output and the mean value of the physicochemical profile. These are combined through a multilayer perceptron trained on the balanced RBP versus non-RBP dataset. In the independent human test set described in the manuscript draft, the HERCULES global score discriminates RBPs from non-RBPs with an AUROC of 0.86.
Mutation Effect Prediction
HERCULES supports mutation analysis by comparing wild-type and mutant sequences through their residue-level profiles. For a given variant (single or multiple substitutions), HERCULES computes a mutation score summarizing the change in RNA-binding propensity, defined as the relative difference between mutant and wild-type residue-level profiles:
On a curated UniProt-derived dataset of experimentally validated RNA-binding-disrupting variants, HERCULES correctly classifies about 87% of deleterious mutations in the held-out test set, including many single-amino acid substitutions.
Web Server Outputs
Given an input protein sequence, the HERCULES web server returns:
- A global RNA-binding propensity score
- A residue-level RNA-binding propensity profile highlighting candidate RNA-binding domains and regions
- An optional mutagenesis heatmap to visualize mutation score along the sequence
Datasets and Validation Summary
HERCULES was trained and systematically benchmarked across three complementary tasks: global RBP classification, residue-level RNA-binding domain localization, and prediction of mutation effects on RNA binding.
For global RNA-binding propensity, the model was trained on a non-redundant set of human RBPs curated for scRAPID-web together with experimentally validated and curated non-RBPs (2-8), using an 80/20 train-test split. On the independent test set, the HERCULES global score discriminates RBPs from non-RBPs with an AUROC of 0.86 and an AUPRC of 0.86. Compared with HydRA (10), HERCULES shows slightly lower binary performance on the human test set, but identifies a larger fraction of annotated RBPs across nine organisms from the RBP2GO database (5) (Figure 2A), reflecting a more permissive and sensitive global classification strategy that supports downstream domain localization.
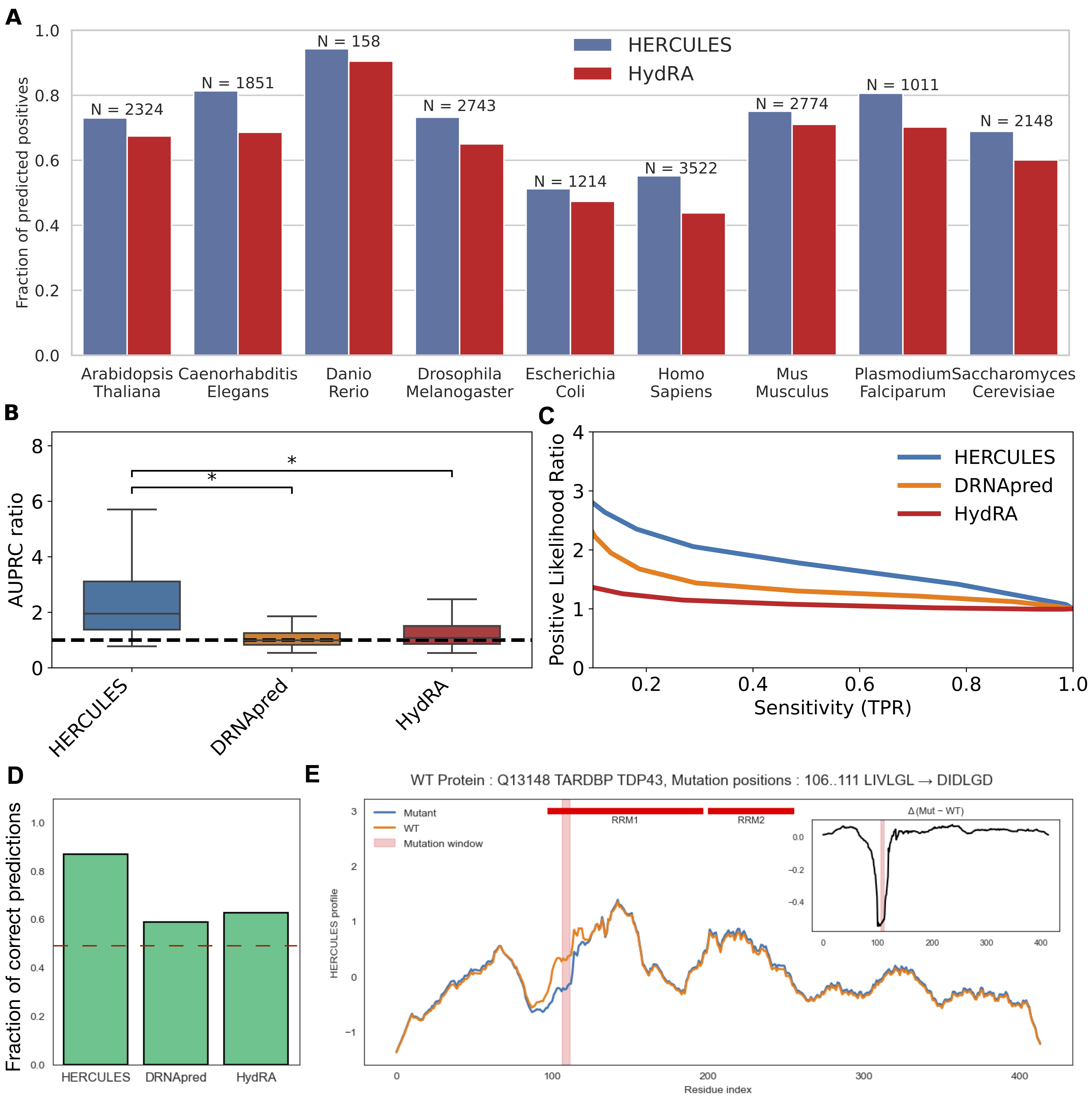
Figure 2. HERCULES performance across tasks. A. Fraction of predicted RBPs across nine organisms using RBPs curated from RBP2GO. Bars show the fraction of proteins classified as RNA-binding by binarized HERCULES and HydRA scores; the number of proteins analyzed in each organism is indicated above each bar. B. Box plots showing the distribution of the Area Under the Precision-Recall Curve (AUPRC), normalized by expected random AUPRC (fraction of RNA-binding residues in each protein), computed over 562 human proteins with Pfam-annotated RNA-binding domains and less than 40% sequence identity with the training set. Colors indicate different prediction algorithms. C. Positive likelihood ratio as a function of sensitivity for the same protein set as panel B. D. Fraction of correctly predicted mutations for HERCULES, DRNApred, and HydRA on the held-out test set. E. HERCULES residue-level RNA-binding propensity profiles for WT (orange) and mutant (blue) sequences around the mutation region. The example protein (TDP-43) carries a multiple amino-acid substitution (positions 106-111, LIVLGL to DIDLGD).
Residue-level performance was evaluated on a non-redundant benchmark of human proteins with Pfam-annotated canonical, non-classical and putative RNA-binding domains (6, 11-13), sharing less than 40% sequence identity with the training set. HERCULES was compared against state-of-the-art sequence-based predictors, including HydRA (10) and DRNApred (14). Across multiple complementary metrics (AUPRC normalized to random expectation, positive likelihood ratio, fold enrichment in top-ranked residues, and top/bottom AUROC analysis), HERCULES consistently outperforms competing methods in identifying bona fide RNA-binding residues (Figure 2B-C). The improvement is particularly pronounced for canonical and non-canonical domains, and remains significant for putative domains, where annotation uncertainty is higher. Notably, HERCULES performance increases with intrinsic disorder content, in contrast to other methods, indicating enhanced sensitivity to disordered RNA-binding regions.
Mutation-effect prediction was assessed using a curated dataset of experimentally validated RNA-binding-disrupting variants derived from UniProt (15), with separation of training and test proteins to prevent information leakage. On the held-out test set, HERCULES correctly classifies approximately 87% of deleterious mutations, outperforming alternative predictors (Figure 2D). Importantly, this performance is achieved using sequence information alone and applies to both single and multiple amino-acid substitutions.
Finally, residue-level predictions were evaluated on a dataset of experimentally resolved protein-RNA complexes from the Protein Data Bank. To address the intrinsic limitation of single-RNA annotations, the benchmark was augmented with AlphaFold3-predicted complexes (16) involving multiple G-quadruplex RNAs, generating an expanded set of RNA-contact residues. Under this augmented evaluation, HERCULES is the only method that shows improved performance, while other predictors exhibit reduced accuracy, suggesting limited generalization beyond their original training annotations. These results demonstrate that HERCULES captures generalizable physicochemical and sequence determinants of RNA binding and maintains robust performance across heterogeneous datasets.
Together, these benchmarks show that HERCULES achieves competitive global RBP classification, residue-level domain localization, and mutation-effect prediction within a unified, fully sequence-based framework.
References
- Brandes,N., Ofer,D., Peleg,Y., Rappoport,N. and Linial,M. (2022) ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics, 38, 2102-2110.
- Fiorentino,J., Armaos,A., Colantoni,A. and Tartaglia,G.G. (2024) Prediction of protein-RNA interactions from single-cell transcriptomic data. Nucleic Acids Res, 52, e31.
- Fiorentino,J., Armaos,A., Montrone,C., Colantoni,A. and Tartaglia,G.G. (2025) scRAPID-web: a web server for predicting protein-RNA interactions from single-cell transcriptomics. bioRxiv, 10.1101/2025.03.12.642785.
- Armaos,A., Colantoni,A., Proietti,G., Rupert,J. and Tartaglia,G.G. (2021) catRAPID omics v2.0: going deeper and wider in the prediction of protein-RNA interactions. Nucleic Acids Res, 49, W72-W79.
- Caudron-Herger,M., Jansen,R.E., Wassmer,E. and Diederichs,S. (2021) RBP2GO: a comprehensive pan-species database on RNA-binding proteins, their interactions and functions. Nucleic Acids Res, 49, D425-D436.
- Castello,A., Fischer,B., Eichelbaum,K., Horos,R., Beckmann,B.M., Strein,C., Davey,N.E., Humphreys,D.T., Preiss,T., Steinmetz,L.M., Krijgsveld,J., and Hentze,M.W. (2012) Insights into RNA Biology from an Atlas of Mammalian mRNA-Binding Proteins. Cell, 149, 1393-1406.
- Monti,M., Fiorentino,J., Miltiadis-Vrachnos,D., Bini,G., Cotrufo,T., Sanchez de Groot,N., Armaos,A. and Tartaglia,G.G. (2025) catGRANULE 2.0: accurate predictions of liquid-liquid phase separating proteins at single amino acid resolution. Genome Biol, 26, 33.
- Peng,X., Wang,X., Guo,Y., Ge,Z., Li,F., Gao,X. and Song,J. (2022) RBP-TSTL is a two-stage transfer learning framework for genome-scale prediction of RNA-binding proteins. Brief Bioinform, 23.
- Klus,P., Bolognesi,B., Agostini,F., Marchese,D., Zanzoni,A. and Tartaglia,G.G. (2014) The cleverSuite approach for protein characterization: predictions of structural properties, solubility, chaperone requirements and RNA-binding abilities. Bioinformatics, 30, 1601-1608.
- Jin,W., Brannan,K.W., Kapeli,K., Park,S.S., Tan,H.Q., Gosztyla,M.L., Mujumdar,M., Ahdout,J., Henroid,B., Rothamel,K., Xiang,J.S., Wong,L., and Yeo,G.W. (2023) HydRA: Deep-learning models for predicting RNA-binding capacity from protein interaction association context and protein sequence. Molecular Cell, 83, 2595-2611.e11.
- Mistry,J., Chuguransky,S., Williams,L., Qureshi,M., Salazar,G.A., Sonnhammer,E.L.L., Tosatto,S.C.E., Paladin,L., Raj,S., Richardson,L.J., et al. (2021) Pfam: The protein families database in 2021. Nucleic Acids Res, 49, D412-D419.
- Kwon,S.C., Yi,H., Eichelbaum,K., Foehr,S., Fischer,B., You,K.T., Castello,A., Krijgsveld,J., Hentze,M.W. and Kim,V.N. (2013) The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol, 20, 1122-1130.
- Baltz,A.G., Munschauer,M., Schwanhausser,B., Vasile,A., Murakawa,Y., Schueler,M., Youngs,N., Penfold-Brown,D., Drew,K., Milek,M., et al. (2012) The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell, 46, 674-690.
- Yan,J. and Kurgan,L. (2017) DRNApred, fast sequence-based method that accurately predicts and discriminates DNA- and RNA-binding residues. Nucleic Acids Res, 45, e84.
- UniProt Consortium (2023) UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res, 51, D523-D531.
- Abramson,J., Adler,J., Dunger,J., Evans,R., Green,T., Pritzel,A., Ronneberger,O., Willmore,L., Ballard,A.J., Bambrick,J., et al. (2024) Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630, 493-500.