Documentation for catGRANULE 2.0 ROBOT
catGRANULE2 ROBOT (Ribonucleoprotein Organization in Biocondensates Organelle Types) is a machine learning method that predicts the propensity of proteins to undergo liquid-liquid phase separation (LLPS), computes a LLPS propensity profile with single amino acid resolution and predicts the effect of mutations of the protein sequence on LLPS propensity (Figure 1).
The method is trained on a dataset of human proteins, composed of 3333 proteins known to be LLPS-prone, collected from several databases [1-5], and 3252 non-LLPS proteins sampled from the human proteome, after the exclusion of the LLPS-prone proteins and their first interactors.
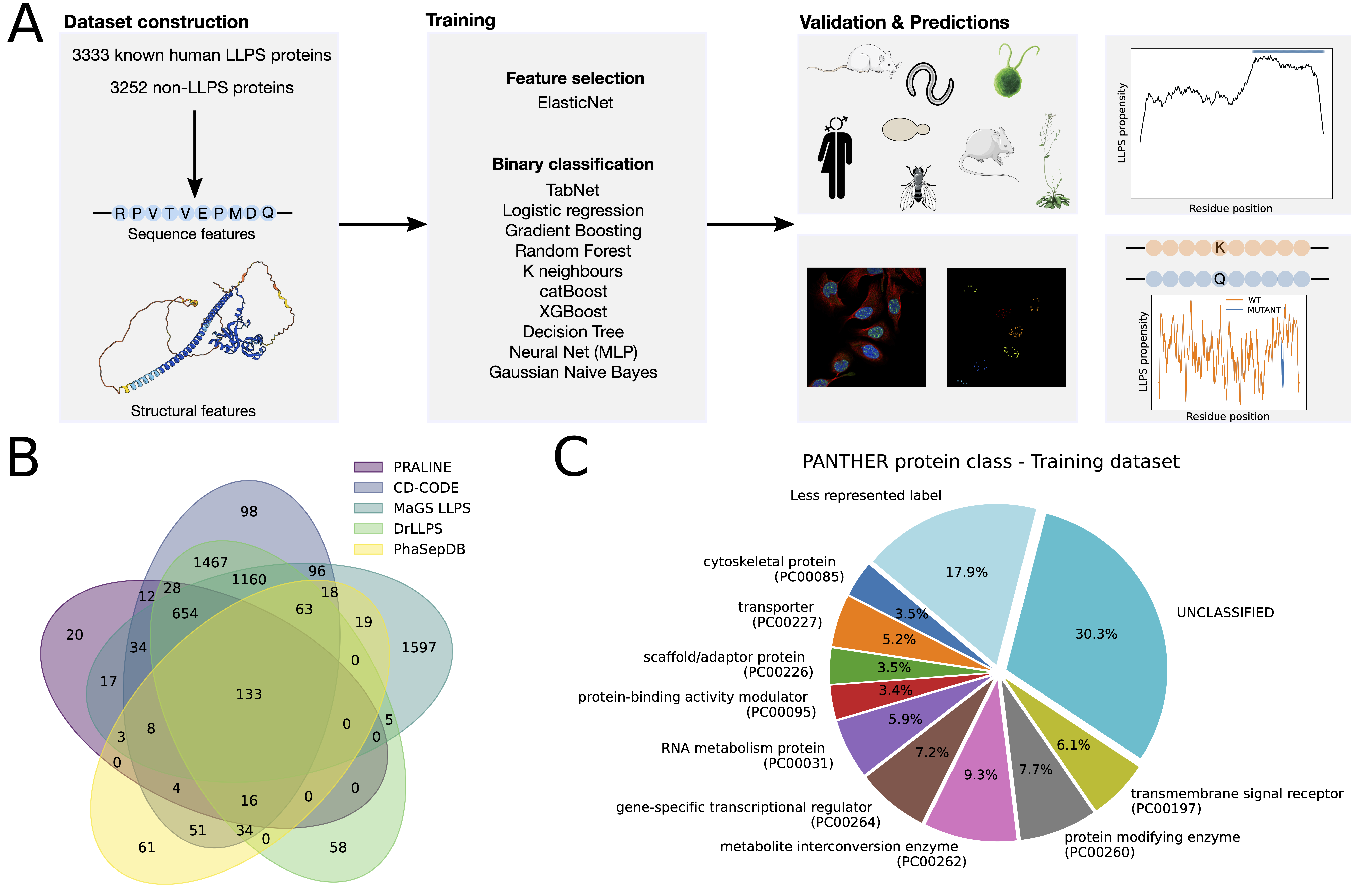
Figure 1. Schematics of the catGRANULE 2.0 ROBOT workflow. A training dataset is constructed consisting of 3333 known human LLPS proteins and 3252 non-LLPS proteins. The proteins are then encoded in a set of 128 features, including sequence-based physico-chemical and Alphafold2-derived structural features.
Each protein in the training dataset is encoded in a vector of 128 features, including 80 sequence-based physico-chemical features as well as 2 additional phenomenological properties [6,7], 28 structural features derived from AlphaFold2 predictions [8] and 18 RNA binding features computed from a compositional similarity analysis of a dataset of experimentally determined RNA-binding peptides [9].
Feature selection is performed via the ElasticNet method, which identifies a set of 28 relevant features. Next, several classifiers are trained using 5-fold cross-validation and their hyperparameters are optimized through a grid search approach, employing the Area Under the Receiver-Operating Characteristic Curve (AUROC) as the scoring function. A Multi-Layer Perceptron (MLP) is selected as the best model, based on its superior performance on an independent test set made of 1422 LLPS-prone proteins and 1376 non-LLPS proteins.
Extensive validation of catGRANULE 2.0 ROBOT and comparison with other state-of-the-art predictors, such as MaGS, PICNIC, PICNIC-GO and PSPHunter [10-12], are carried out on the independent test set, on known LLPS proteins from different species collected from the DrLLPS database [3] and on antibody-based immunofluorescence confocal microscopy images obtained from the Human Protein Atlas [13] (Figure 2).
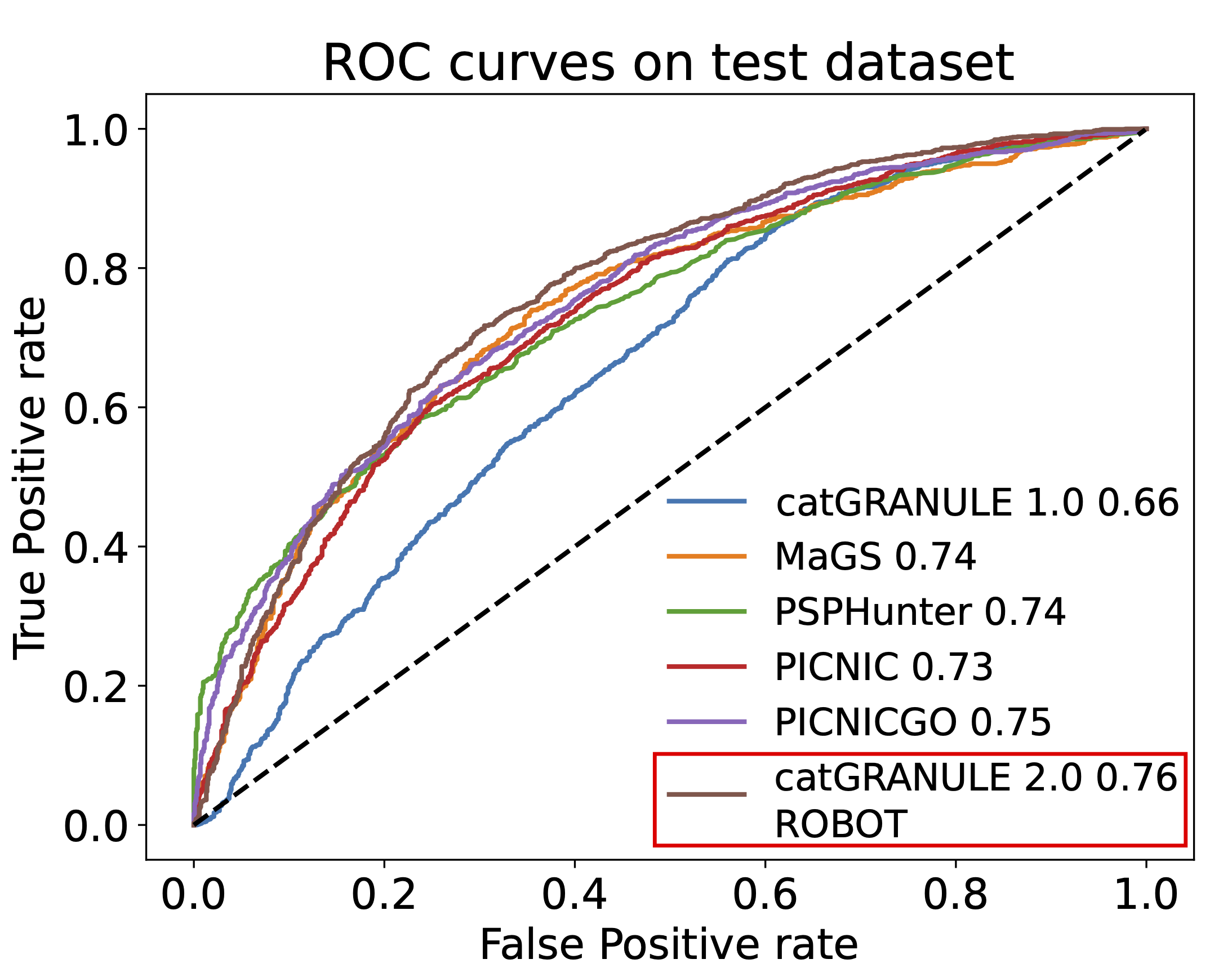
Figure 2. Performances on the test dataset, for catGRANULE 2.0 ROBOT and other LLPS prediction algorithms.
Beyond the computation of a LLPS propensity score, which is the prediction probability of the trained MLP model on the selected features, catGRANULE2 ROBOT computes a LLPS propensity profile of a protein with single amino acid resolution and it accurately predicts the effect of single and multiple amino acid mutations on LLPS propensity.
The LLPS propensity profile of a protein is built by applying a sliding window to the protein sequence, and predicting the LLPS propensity score using the trained model. The computation of the profiles is validated on a set of approximately 250 proteins from different species, collected from the PhaSepDB database [2], for which the sequence region driving LLPS was experimentally determined. The optimal size of the sliding window, which is 23 amino acids, is found by optimizing the performance against the experimental data.
For the computation of the LLPS propensity profiles and mutation scoring, catGRANULE 2 ROBOT employs a Random Forest classifier trained using only the set of physico-chemical ROBOT properties. This choice is supported by the superior performance of this classifier compared to the others in scoring LLPS propensity profiles, the faster computational time compared to the full MLP model trained also on structural features, which makes it suitable for application to deep mutational scanning of proteins, and the poor performance of AlphaFold2 in predicting the structure of proteins under single point mutations, previously known from different studies [14].
To predict the effect of mutations on LLPS propensity, we define a mutation score based on the profiles of the wild-type (WT) and mutant proteins in the following way:
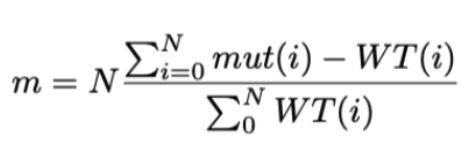
To test the capability of catGRANULE2 ROBOT in predicting the positive or negative effect of mutations on LLPS propensity, we compiled a curated list of 24 single and multiple amino acid mutations of 9 proteins that increase or decrease their LLPS propensity, and we found that catGRANULE2 ROBOT outperforms previous algorithms in correctly identifying the effect of the mutation on LLPS. Finally, as a further validation we showed that catGRANULE2 ROBOT accurately predicts the effect of single and double point mutations on LLPS from a mutational scanning of the prion-like domain of TDP-43 [15].
The accuracy of the model on the test set reaches 0.76 of AUROC, leading this algorithm to be the most reliable at the state of the art.
References:
- Vandelli A, Arnal Segura M, Monti M, Fiorentino J, Broglia L, Colantoni A, et al. The PRALINE database: protein and Rna humAn singLe nucleotIde variaNts in condEnsates. Bioinformatics [Internet]. 2023;39. Available from: link
- You K, Huang Q, Yu C, Shen B, Sevilla C, Shi M, et al. PhaSepDB: a database of liquid-liquid phase separation related proteins. Nucleic Acids Res. 2019;48:D354-9.
- Ning W, Guo Y, Lin S, Mei B, Wu Y, Jiang P, et al. DrLLPS: a data resource of liquid-liquid phase separation in eukaryotes. Nucleic Acids Res. 2019;48:D288-95.
- Wang X, Zhou X, Yan Q, Liao S, Tang W, Xu P, et al. LLPSDB v2.0: an updated database of proteins undergoing liquid-liquid phase separation in vitro. Bioinformatics. 2022;38:2010-4.
- Kuechler ER, Huang A, Bui JM, Mayor T, Gsponer J. Comparison of Biomolecular Condensate Localization and Protein Phase Separation Predictors. Biomolecules [Internet]. 2023;13. Available from: link
- Klus P, Bolognesi B, Agostini F, Marchese D, Zanzoni A, Tartaglia GG. The cleverSuite approach for protein characterization: predictions of structural properties, solubility, chaperone requirements and RNA-binding abilities. Bioinformatics. 2014;30:1601-8.
- Bolognesi B, Lorenzo Gotor N, Dhar R, Cirillo D, Baldrighi M, Tartaglia GG, et al. A Concentration-Dependent Liquid Phase Separation Can Cause Toxicity upon Increased Protein Expression. Cell Rep. 2016;16:222-31.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583-9.
- Castello A, Fischer B, Frese CK, Horos R, Alleaume A-M, Foehr S, et al. Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol Cell. 2016;63:696-710.
- Kuechler ER, Budzyńska PM, Bernardini JP, Gsponer J, Mayor T. Distinct Features of Stress Granule Proteins Predict Localization in Membraneless Organelles. J Mol Biol. 2020 Mar 27;432(7):2349-2368. doi: 10.1016/j.jmb.2020.02.020. Epub 2020 Feb 24. PMID: 32105731.
- Hadarovich A, Singh HR, Ghosh S, Rostam N, Hyman AA, Toth-Petroczy A. PICNIC accurately predicts condensate-forming proteins regardless of their structural disorder across organisms [Internet]. bioRxiv. 2023 [cited 2024 Jun 4]. p. 2023.06.01.543229. Available from: link
- Sun J, Qu J, Zhao C, Zhang X, Liu X, Wang J, et al. Precise prediction of phase-separation key residues by machine learning. Nat Commun. 2024;15:2662.
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419.
- Buel GR, Walters KJ. Can AlphaFold2 predict the impact of missense mutations on structure? Nat Struct Mol Biol. 2022;29:1-2.
- Bolognesi B, Faure AJ, Seuma M, Schmiedel JM, Tartaglia GG, Lehner B. The mutational landscape of a prion-like domain. Nat Commun. 2019;10:4162.